Performance summary workflow (targeted bed)¶
PerformanceSummaryTargeted · 1 contributor · 1 version
No documentation was provided: contribute one
Quickstart¶
from janis_bioinformatics.tools.pmac.performanceSummaryTargetedWorkflow import PerformanceSummaryTargeted_0_1_0 wf = WorkflowBuilder("myworkflow") wf.step( "performancesummarytargeted_step", PerformanceSummaryTargeted_0_1_0( bam=None, genecoverage_bed=None, region_bed=None, sample_name=None, genome_file=None, ) ) wf.output("out", source=performancesummarytargeted_step.out) wf.output("geneFileOut", source=performancesummarytargeted_step.geneFileOut) wf.output("regionFileOut", source=performancesummarytargeted_step.regionFileOut)
OR
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
- Generate user input files for PerformanceSummaryTargeted:
# user inputs
janis inputs PerformanceSummaryTargeted > inputs.yaml
inputs.yaml
bam: bam.bam
genecoverage_bed: genecoverage_bed.bed
genome_file: genome_file.txt
region_bed: region_bed.bed
sample_name: <value>
- Run PerformanceSummaryTargeted with:
janis run [...run options] \
--inputs inputs.yaml \
PerformanceSummaryTargeted
Information¶
URL: No URL to the documentation was provided
| ID: | PerformanceSummaryTargeted |
|---|---|
| URL: | No URL to the documentation was provided |
| Versions: | v0.1.0 |
| Authors: | Jiaan Yu |
| Citations: | |
| Created: | 2020-04-28 |
| Updated: | 2020-10-05 |
Outputs¶
| name | type | documentation |
|---|---|---|
| out | csv | |
| geneFileOut | TextFile | |
| regionFileOut | TextFile |
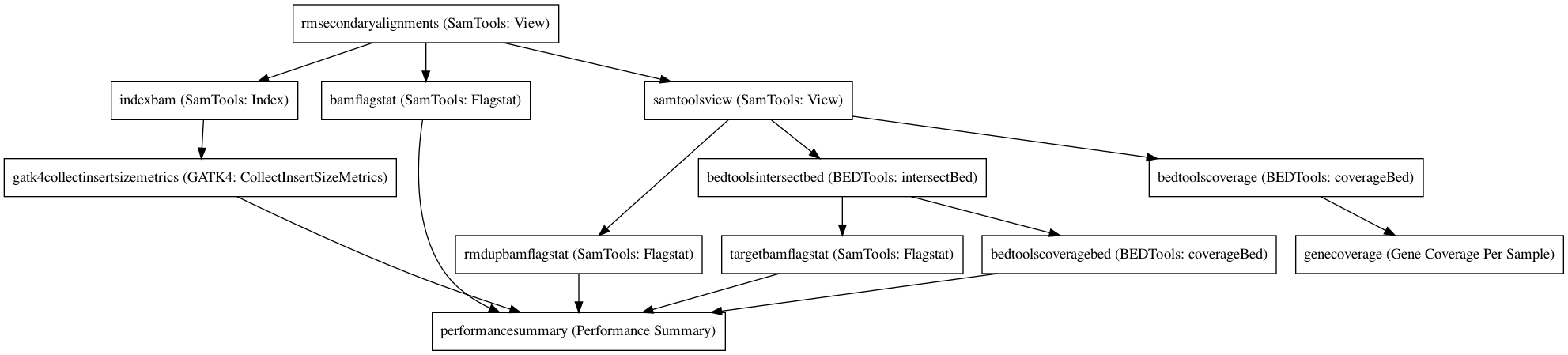
Workflow¶
Embedded Tools¶
| SamTools: View | SamToolsView/1.9.0 |
| SamTools: Index | SamToolsIndex/1.9.0 |
| GATK4: CollectInsertSizeMetrics | Gatk4CollectInsertSizeMetrics/4.1.2.0 |
| SamTools: Flagstat | SamToolsFlagstat/1.9.0 |
| BEDTools: intersectBed | bedtoolsintersectBed/v2.29.2 |
| BEDTools: coverageBed | bedtoolsCoverageBed/v2.29.2 |
| Performance Summary | performanceSummary/0.0.7 |
| Gene Coverage Per Sample | geneCoveragePerSample/0.0.8 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| bam | IndexedBam | |
| genecoverage_bed | bed | |
| region_bed | bed | |
| sample_name | String | |
| genome_file | TextFile | |
| rmsecondaryalignments_doNotOutputAlignmentsWithBitsSet | Optional<String> | Do not output alignments with any bits set in INT present in the FLAG field. INT can be specified in hex by beginning with `0x’ (i.e. /^0x[0-9A-F]+/) or in octal by beginning with `0’ (i.e. /^0[0-7]+/) [0]. |
| samtoolsview_doNotOutputAlignmentsWithBitsSet | Optional<String> | Do not output alignments with any bits set in INT present in the FLAG field. INT can be specified in hex by beginning with `0x’ (i.e. /^0x[0-9A-F]+/) or in octal by beginning with `0’ (i.e. /^0[0-7]+/) [0]. |
| bedtoolsintersectbed_sorted | Optional<Boolean> | Use the ‘chromsweep’ algorithm for sorted (-k1,1 -k2,2n) input. |
| bedtoolscoveragebed_sorted | Optional<Boolean> | Use the ‘chromsweep’ algorithm for sorted (-k1,1 -k2,2n) input. |
| bedtoolscoveragebed_histogram | Optional<Boolean> | Report a histogram of coverage for each feature in A as well as a summary histogram for _all_ features in A. Output (tab delimited) after each feature in A: 1) depth 2) # bases at depth 3) size of A 4) % of A at depth. |
| bedtoolscoverage_sorted | Optional<Boolean> | Use the ‘chromsweep’ algorithm for sorted (-k1,1 -k2,2n) input. |
| bedtoolscoverage_histogram | Optional<Boolean> | Report a histogram of coverage for each feature in A as well as a summary histogram for _all_ features in A. Output (tab delimited) after each feature in A: 1) depth 2) # bases at depth 3) size of A 4) % of A at depth. |
Workflow Description Language¶
version development
import "tools/SamToolsView_1_9_0.wdl" as S
import "tools/SamToolsIndex_1_9_0.wdl" as S2
import "tools/Gatk4CollectInsertSizeMetrics_4_1_2_0.wdl" as G
import "tools/SamToolsFlagstat_1_9_0.wdl" as S3
import "tools/bedtoolsintersectBed_v2_29_2.wdl" as B
import "tools/bedtoolsCoverageBed_v2_29_2.wdl" as B2
import "tools/performanceSummary_0_0_7.wdl" as P
import "tools/geneCoveragePerSample_0_0_8.wdl" as G2
workflow PerformanceSummaryTargeted {
input {
File bam
File bam_bai
File genecoverage_bed
File region_bed
String sample_name
File genome_file
String? rmsecondaryalignments_doNotOutputAlignmentsWithBitsSet = "0x100"
String? samtoolsview_doNotOutputAlignmentsWithBitsSet = "0x400"
Boolean? bedtoolsintersectbed_sorted = true
Boolean? bedtoolscoveragebed_sorted = true
Boolean? bedtoolscoveragebed_histogram = true
Boolean? bedtoolscoverage_sorted = true
Boolean? bedtoolscoverage_histogram = true
}
call S.SamToolsView as rmsecondaryalignments {
input:
doNotOutputAlignmentsWithBitsSet=select_first([rmsecondaryalignments_doNotOutputAlignmentsWithBitsSet, "0x100"]),
sam=bam
}
call S2.SamToolsIndex as indexbam {
input:
bam=rmsecondaryalignments.out
}
call G.Gatk4CollectInsertSizeMetrics as gatk4collectinsertsizemetrics {
input:
bam=indexbam.out,
bam_bai=indexbam.out_bai
}
call S3.SamToolsFlagstat as bamflagstat {
input:
bam=rmsecondaryalignments.out
}
call S.SamToolsView as samtoolsview {
input:
doNotOutputAlignmentsWithBitsSet=select_first([samtoolsview_doNotOutputAlignmentsWithBitsSet, "0x400"]),
sam=rmsecondaryalignments.out
}
call S3.SamToolsFlagstat as rmdupbamflagstat {
input:
bam=samtoolsview.out
}
call B.bedtoolsintersectBed as bedtoolsintersectbed {
input:
genome=genome_file,
sorted=select_first([bedtoolsintersectbed_sorted, true]),
inputABam=samtoolsview.out,
inputBBed=[region_bed]
}
call S3.SamToolsFlagstat as targetbamflagstat {
input:
bam=bedtoolsintersectbed.out
}
call B2.bedtoolsCoverageBed as bedtoolscoveragebed {
input:
genome=genome_file,
sorted=select_first([bedtoolscoveragebed_sorted, true]),
inputABed=region_bed,
inputBBam=bedtoolsintersectbed.out,
histogram=select_first([bedtoolscoveragebed_histogram, true])
}
call P.performanceSummary as performancesummary {
input:
flagstat=bamflagstat.out,
collectInsertSizeMetrics=gatk4collectinsertsizemetrics.out,
coverage=bedtoolscoveragebed.out,
outputPrefix=sample_name,
targetFlagstat=targetbamflagstat.out,
rmdupFlagstat=rmdupbamflagstat.out
}
call B2.bedtoolsCoverageBed as bedtoolscoverage {
input:
genome=genome_file,
sorted=select_first([bedtoolscoverage_sorted, true]),
inputABed=genecoverage_bed,
inputBBam=samtoolsview.out,
histogram=select_first([bedtoolscoverage_histogram, true])
}
call G2.geneCoveragePerSample as genecoverage {
input:
sampleName=sample_name,
bedtoolsOutputPath=bedtoolscoverage.out
}
output {
File out = performancesummary.out
File geneFileOut = genecoverage.geneFileOut
File regionFileOut = genecoverage.regionFileOut
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: Performance summary workflow (targeted bed)
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
- class: MultipleInputFeatureRequirement
inputs:
- id: bam
type: File
secondaryFiles:
- pattern: .bai
- id: genecoverage_bed
type: File
- id: region_bed
type: File
- id: sample_name
type: string
- id: genome_file
type: File
- id: rmsecondaryalignments_doNotOutputAlignmentsWithBitsSet
doc: |-
Do not output alignments with any bits set in INT present in the FLAG field. INT can be specified in hex by beginning with `0x' (i.e. /^0x[0-9A-F]+/) or in octal by beginning with `0' (i.e. /^0[0-7]+/) [0].
type: string
default: '0x100'
- id: samtoolsview_doNotOutputAlignmentsWithBitsSet
doc: |-
Do not output alignments with any bits set in INT present in the FLAG field. INT can be specified in hex by beginning with `0x' (i.e. /^0x[0-9A-F]+/) or in octal by beginning with `0' (i.e. /^0[0-7]+/) [0].
type: string
default: '0x400'
- id: bedtoolsintersectbed_sorted
doc: Use the 'chromsweep' algorithm for sorted (-k1,1 -k2,2n) input.
type: boolean
default: true
- id: bedtoolscoveragebed_sorted
doc: Use the 'chromsweep' algorithm for sorted (-k1,1 -k2,2n) input.
type: boolean
default: true
- id: bedtoolscoveragebed_histogram
doc: |-
Report a histogram of coverage for each feature in A as well as a summary histogram for _all_ features in A. Output (tab delimited) after each feature in A: 1) depth 2) # bases at depth 3) size of A 4) % of A at depth.
type: boolean
default: true
- id: bedtoolscoverage_sorted
doc: Use the 'chromsweep' algorithm for sorted (-k1,1 -k2,2n) input.
type: boolean
default: true
- id: bedtoolscoverage_histogram
doc: |-
Report a histogram of coverage for each feature in A as well as a summary histogram for _all_ features in A. Output (tab delimited) after each feature in A: 1) depth 2) # bases at depth 3) size of A 4) % of A at depth.
type: boolean
default: true
outputs:
- id: out
type: File
outputSource: performancesummary/out
- id: geneFileOut
type: File
outputSource: genecoverage/geneFileOut
- id: regionFileOut
type: File
outputSource: genecoverage/regionFileOut
steps:
- id: rmsecondaryalignments
label: 'SamTools: View'
in:
- id: doNotOutputAlignmentsWithBitsSet
source: rmsecondaryalignments_doNotOutputAlignmentsWithBitsSet
- id: sam
source: bam
run: tools/SamToolsView_1_9_0.cwl
out:
- id: out
- id: indexbam
label: 'SamTools: Index'
in:
- id: bam
source: rmsecondaryalignments/out
run: tools/SamToolsIndex_1_9_0.cwl
out:
- id: out
- id: gatk4collectinsertsizemetrics
label: 'GATK4: CollectInsertSizeMetrics'
in:
- id: bam
source: indexbam/out
run: tools/Gatk4CollectInsertSizeMetrics_4_1_2_0.cwl
out:
- id: out
- id: outHistogram
- id: bamflagstat
label: 'SamTools: Flagstat'
in:
- id: bam
source: rmsecondaryalignments/out
run: tools/SamToolsFlagstat_1_9_0.cwl
out:
- id: out
- id: samtoolsview
label: 'SamTools: View'
in:
- id: doNotOutputAlignmentsWithBitsSet
source: samtoolsview_doNotOutputAlignmentsWithBitsSet
- id: sam
source: rmsecondaryalignments/out
run: tools/SamToolsView_1_9_0.cwl
out:
- id: out
- id: rmdupbamflagstat
label: 'SamTools: Flagstat'
in:
- id: bam
source: samtoolsview/out
run: tools/SamToolsFlagstat_1_9_0.cwl
out:
- id: out
- id: bedtoolsintersectbed
label: 'BEDTools: intersectBed'
in:
- id: genome
source: genome_file
- id: sorted
source: bedtoolsintersectbed_sorted
- id: inputABam
source: samtoolsview/out
- id: inputBBed
source:
- region_bed
linkMerge: merge_nested
run: tools/bedtoolsintersectBed_v2_29_2.cwl
out:
- id: out
- id: targetbamflagstat
label: 'SamTools: Flagstat'
in:
- id: bam
source: bedtoolsintersectbed/out
run: tools/SamToolsFlagstat_1_9_0.cwl
out:
- id: out
- id: bedtoolscoveragebed
label: 'BEDTools: coverageBed'
in:
- id: genome
source: genome_file
- id: sorted
source: bedtoolscoveragebed_sorted
- id: inputABed
source: region_bed
- id: inputBBam
source: bedtoolsintersectbed/out
- id: histogram
source: bedtoolscoveragebed_histogram
run: tools/bedtoolsCoverageBed_v2_29_2.cwl
out:
- id: out
- id: performancesummary
label: Performance Summary
in:
- id: flagstat
source: bamflagstat/out
- id: collectInsertSizeMetrics
source: gatk4collectinsertsizemetrics/out
- id: coverage
source: bedtoolscoveragebed/out
- id: outputPrefix
source: sample_name
- id: targetFlagstat
source: targetbamflagstat/out
- id: rmdupFlagstat
source: rmdupbamflagstat/out
run: tools/performanceSummary_0_0_7.cwl
out:
- id: out
- id: bedtoolscoverage
label: 'BEDTools: coverageBed'
in:
- id: genome
source: genome_file
- id: sorted
source: bedtoolscoverage_sorted
- id: inputABed
source: genecoverage_bed
- id: inputBBam
source: samtoolsview/out
- id: histogram
source: bedtoolscoverage_histogram
run: tools/bedtoolsCoverageBed_v2_29_2.cwl
out:
- id: out
- id: genecoverage
label: Gene Coverage Per Sample
in:
- id: sampleName
source: sample_name
- id: bedtoolsOutputPath
source: bedtoolscoverage/out
run: tools/geneCoveragePerSample_0_0_8.cwl
out:
- id: geneFileOut
- id: regionFileOut
id: PerformanceSummaryTargeted